Java 朴素贝叶斯分类器、SVM(5行代码)实现乳腺癌分类
2021-03-02 16:27
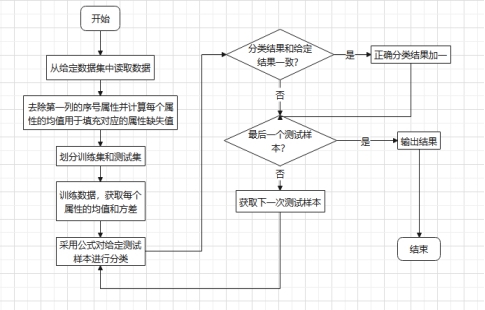
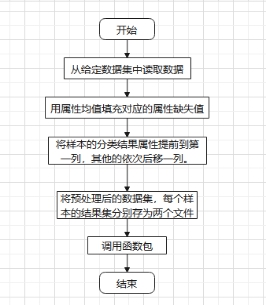
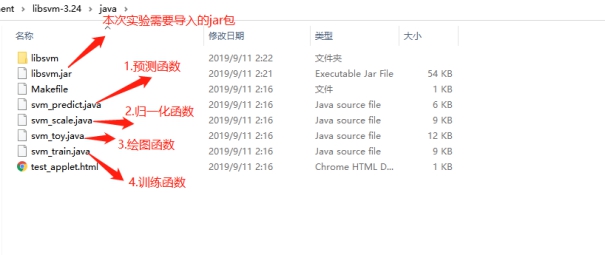
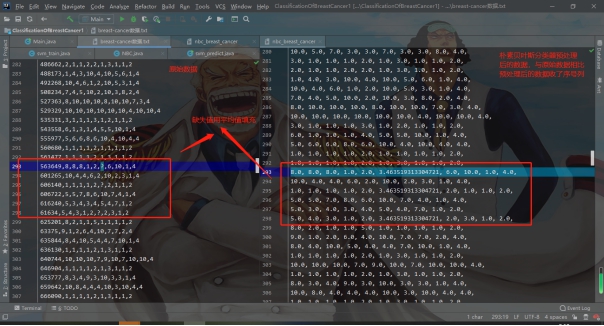
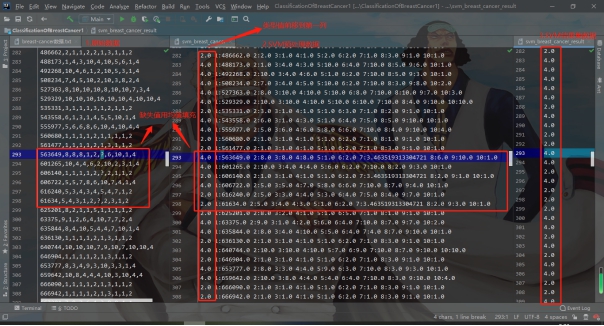
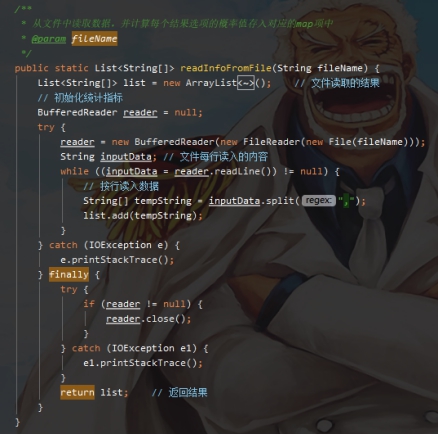
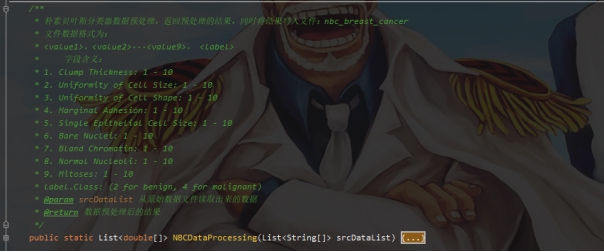
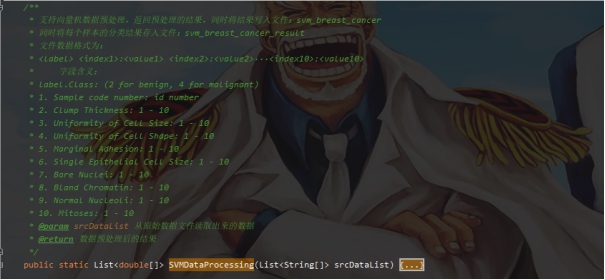
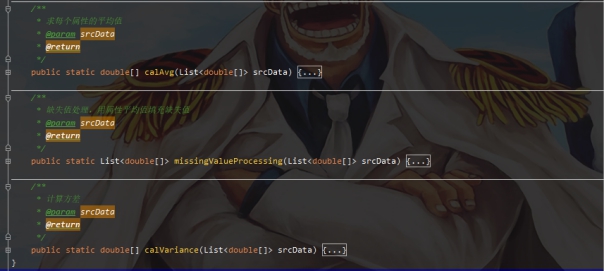
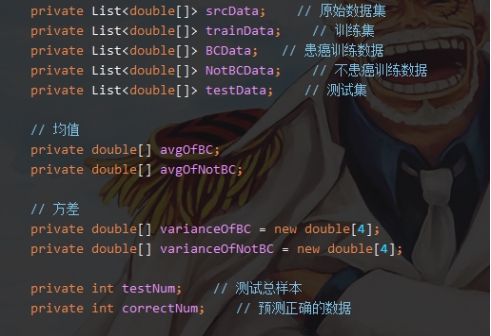
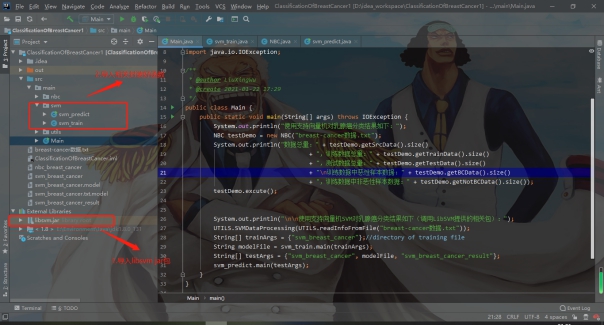
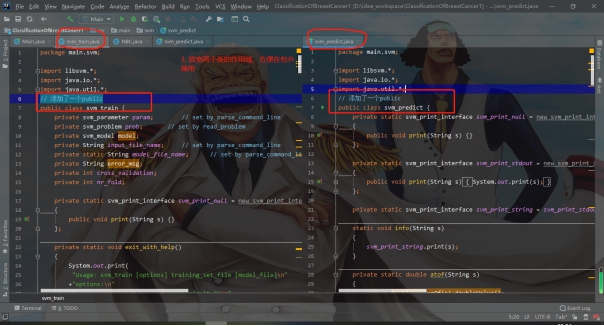
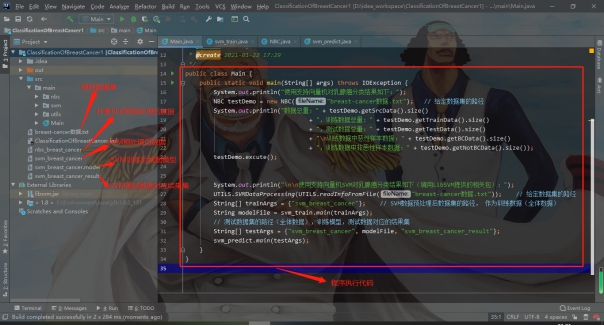
标签:报告 密度 类别 详细 函数调用 can 文件中 svm 实验环境 实验源码:https://gitee.com/LiuXingwu/sharing 某研究获取了若干乳腺癌诊断数据,存放于breast cancer数据.txt 中。每个样本第一个数值为ID,随后10列为十个属性值,最后一列为分类(2代表良性,4代表恶性) 缺失属性用?表示。根据实验算法的不同自行从数据集中选出训练样本和测试样本。 实验要求如下: 1)进行数据预处理,填充缺失值,说明预处理的方法。 2)分别使用支持向量机和朴素贝叶斯分类器进行分类,并比较两种方法在测试样本上的准确率。 3)实验报告中说明训练样本和测试样本如何选择。 4)为了使得数据适应所选择的分类器,要进行适当的数据预处理。 breast cancer数据.txt部分截图: 朴素贝叶斯分类器:样本中给定的十一个属性值中,第一列是样本的序号,这个序号对本次的分类而言属于无关变量,所以在数据初始化时去除这个属性。朴素贝叶斯分类器对于数据的输入格式没有过大的要求,因此在去除样本序号属性后可以直接用于程序分析。在数据预处理期间,先将属性值中出现“?”(即缺失值),用0来替代,在读入全部数据后,计算出每个属性的平均值,用于填充缺失的属性值。对于给定的数据集,采用前2/3作为程序训练样本(大概460多个样本),其余1/3样本作为程序的测试集(大概230多个样本)。在训练过程中,将训练集按照类别的不同(样本集的最后一个属性:2表示非恶性,4表示恶性)划分为两类,再计算出每个属性的平均值和方差。对于每个测试样本的每个属性,可以通过密度函数(正态分布)来获取它对应的概率值。通过朴素贝叶斯公示计算样本属于两个不同类的概率各是多少。得出预测结果后再和测试样本原先给定的分类结果进行比较,如果一致则正确分类的样本数量加一,最后通过正确分类的样本数量和测试样本的数量比值得出程序分类的正确率。 支持向量机(SVM):该方法的实现主要通过调用LibSVM中给定的函数包实现(5行代码实现SVM分类)。因此需要的的就是将给定的数据集按照符合调用包需要的格式整理即可。其中对于样本数据的缺失值也采用属性平均值进行填充。符合LibSVM给定数据输入的格式为: 其中每个字段含义: ? label.Class: (2 for benign, 4 for malignant) Sample code number: id number Clump Thickness: 1 - 10 Uniformity of Cell Size: 1 - 10 Uniformity of Cell Shape: 1 - 10 Marginal Adhesion: 1 - 10 Single Epithelial Cell Size: 1 - 10 Bare Nuclei: 1 - 10 Bland Chromatin: 1 - 10 Normal Nucleoli: 1 - 10 Mitoses: 1 - 10 要获取目标数据格式,只需将给定的数据集最后一列提前到第一列,其余几列依次后移一列即可。由于还需要一个样本集的结果列表,因此在数据预处理期间将每个样本的结果独立放入一个文件中,以备后面函数调用。最后根据需要对调用的函数进行细微调整即可(具体调整下面会详细说明)。 朴素贝叶斯分类器程序流程: SVM程序流程: 使用平台和编程语言介绍:使用的实验平台是IntelliJ IDEA 2019.3.3 x64。使用的编程语言是java。Java是属于面向对象编程语言,能很好地将抽象的问题实例化。 调用的第三方函数包介绍:LIBSVM是台湾大学林智仁(Lin Chih-Jen)教授等开发设计的一个简单、易于使用和快速有效的SVM模式识别与回归的软件包,他不但提供了编译好的可在Windows系列系统的执行文件,还提供了源代码,方便改进、修改以及在其它操作系统上应用;该软件对SVM所涉及的参数调节相对比较少,提供了很多的默认参数,利用这些默认参数可以解决很多问题;并提供了交互检验(Cross Validation)的功能。该软件可以解决C-SVM、ν-SVM、ε-SVR和ν-SVR等问题,包括基于一对一算法的多类模式识别问题。 LibSVM官方地址:https://www.csie.ntu.edu.tw/~cjlin/libsvm/ LibSVM基本使用:https://blog.csdn.net/yushupan/article/details/78998128 网站首页: 下载最新版本为3.24 (目前): 本次实验项目需要导入的除了jar外还有1、4: 数据预处理:对给定的数据集中存在的缺失值,现将缺失值置为0,然后利用其对应属性的均值填充缺失值。对于朴素贝叶斯分类器分类,去除原始数据中的第一列(序号属性)。对于支持向量机分类,将样本类别属性提前到第一列(循环右移一列),同时将样本的结果集另存一个文件夹中。 封装的工具类: 从原始数据中读取数据: 朴素贝叶斯分类器数据预处理: SVM数据预处理: 求方差、均值、属性值填充: 朴素贝叶斯分类器: 数据变量: 获取训练集测试集、训练参数以及单属性求概率: 程序执行: SVM: 实验结果: 从实验结果来看,朴素贝叶斯分类器的正确率为97%,而SVM的准确率可以达到100%。总体上,两类的分类效果都比较理想。 对于朴素贝叶斯份分类器的结果出现一定的误差我觉得可能是密度函数方面的选取有关,实验中我采用的是求出各个属性的均值和方差然后将对应的正态分布转成标准正态分布的方式来求对应的密度值的,这点有可能会影响最后的结果。也可能是采用平均值填充缺失值造成一定的影响。 对于SVM的分类准确度,我个人是比较惊讶的。属于出乎意料之外,又在情理之中。所谓出乎意料,是因为即使是调用库函数,但我也没想过可以做到100%的精确度,可能是因为我在做完数据预处理后,将全部样本用于训练模型,同时也作为测试样本的原因吧。在情理之中是因为SVM本来就对二类分类的高效性,虽然样本数据近700个,但仍属于小规模样本,所以很好地契合了SVM分类的优势。 从第5章开始学习朴素贝叶斯开始,做了相关的作业,做了两次实验(一次是朴素贝叶斯分类器的实现、一次是鸢尾花分类),在加上这次的乳腺癌诊断。对于朴素贝叶斯分类的算法流程已经相对熟悉了。个人觉得影响分类结果的两大因素为:缺失值的处理(涉及到相关参数的训练)和密度函数的选取(涉及到求取最后的分类概率)。因为各个属性的相互独立,在这个实验中基本是可以保证的。 对于SVM从一开始学习就只是简单了解的其实现的流程和大致适用的情形。对于具体的实现也是一直存有疑虑的。对于之前使用鸢尾花分类和这次的乳腺癌诊断都采用的是调用库函数,其中需要注意的就是按照函数的要求做好数据预处理而已。 结合之前的鸢尾花分类实验,我觉得朴素贝叶斯分类器实现数据分类确实相对简单,而且执行速度也很理想,但始终没法达到特别高了准确率(这次的97%确实出乎意料)。而SVM处理二类划分的效果比之前鸢尾花的三类划分确实好上不少,虽然具体实现较为复杂,但相对于朴素贝叶斯分类器而言SVM有准确率的保证。所以在有库函数的前提下,采用SVM来处理数据不失为更好的选择。 实验源码:https://gitee.com/LiuXingwu/sharing Java 朴素贝叶斯分类器、SVM(5行代码)实现乳腺癌分类 标签:报告 密度 类别 详细 函数调用 can 文件中 svm 实验环境 原文地址:https://www.cnblogs.com/Liu-xing-wu/p/14337600.htmlJava实现乳腺癌诊断(分类)实验总结
朴素贝叶斯分类器、SVM(5行代码实现)
1.问题描述
2. 实验目的
1、加强对朴素贝叶斯分类算法和支持向量机工作过程的理解;
2、锻炼分析问题、解决问题并动手实践的能力。
3. 方法与步骤
3.1 方法概述
3.2 算法描述
4. 实验与结果分析
4.1 实验环境与参数设置

4.2 实验数据
4.3 实验过程与结果
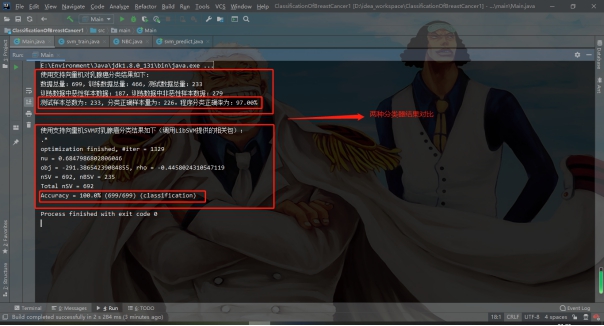
4.4 结果分析
5. 总结与体会
文章标题:Java 朴素贝叶斯分类器、SVM(5行代码)实现乳腺癌分类
文章链接:http://soscw.com/index.php/essay/59103.html